
DNA methylation is a crucial epigenetic mark in regulating various biological processes, including heterochromatin formation, gene imprinting, X chromosome inactivation and transposon element silencing. In addition, reduced DNA methylation is often observed in cancer cells.
In mammals, DNA methylation occurs mainly in CpG islands (5mC), which is mediated by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A and DNMT3B. About 80 % CpGs in human genome are methylated and the methylation level is highly dynamic in early embryonic development. Yet it remains unclear how DNA methylation patterns in different genomic regions, especially intergenic regions (in between genes), are established and maintained, and how histone modifications are involved in this process. One well-known example is the recognition of unmethylated histone H3K4 by the ADD domain of DNMT3A which could further stimulate DNMT3A activity, while methylated histone H3K4 tails could not do so. Such mechanism is believed to protect active promoters and enhancers from DNA methylation. In addition, gene body H3K36me3 has been demonstrated to play a critical role in recruiting DNMT3B through its PWWP domain, which mediates genic DNA methylation and suppresses spurious error-prone transcription. However, the mechanism by which DNA methylation is regulated at intergenic regions remains unknown.
Recently, a collaborative study led by Prof. Fei Lan’s team together with Dr. Hongjie Shen’s and Dr. Feizhen Wu’s groups, all from Institutes of Biomedical Sciences of Fudan University, was published as a Letter to Editor in Protein & Cell. The study was entitled as “DNMT3A Reads and Connects H3K36me2 to DNA Methylation”, which revealed a previously under-appreciated function of H3K36me2 in regulating DNMT3A activity and recruitment primarily in the intergenic regions. The research team also demonstrated the biological impact of the mechanism in promoting multiple myeloma (an aggressive type of plasm cell cancer) cell growth.
H3K36me2 is an abundant histone modification in mammals, and is widely found in both intergenic and genic regions, but depleted in promoters. Histone profiling reveals that its abundance can reach 10%-20%, far greater than that of H3K36me3 whose abundance is only around 4-5%. Yet the biological functions of H3K36me2 remain elusive. By whole genome sequencing, the researchers observed a strong correlation between H3K36me2 and 5mC (R = 0.81) especially in the intergenic regions and the immediate promoter upstream regions, which is much higher than that between H3K36me3 and 5mC (R = 0.63). Such strong correlation was surprising when Lan team initially observed, and the team soon came up with a hypothesis that H3K36me2 might play a role in directing DNA methylation in a broader area of the genome. Following such hypothesis, Lan’s team quickly identified a specific interaction between H3K36me2 and the PWWP of DNMT3A, and importantly, DNMT3A activity could be substantially enhanced by the interaction. It is one of a few crosstalking mechanisms between histone modifications and DNA methylation.
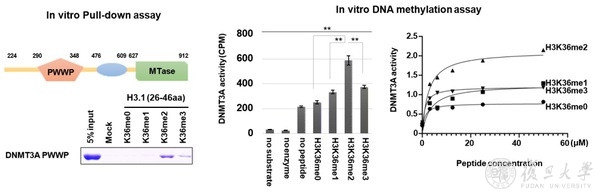
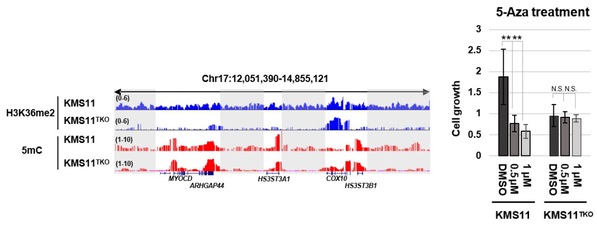
The researchers further explored the biological and pathological significance of such crosstalk. H3K36me2 is mainly catalyzed by the NSD (nuclear receptor SET domain-containing) family of histone lysine methyltransferases, also known as MMSET or WHSC1. NSD gene mutations are often found in various diseases. For instance, the massive overexpression of NSD2 and genome-wide gain of H3K36me2 caused by the chromosomal translocation event, T(4;14), an epigenetics driver mutation occurring in ~20% multiple myeloma. The researchers took the advantage of a well-characterized multiple myeloma cell model, KMS11, which contains the T(4;14) translocation and is a great model here to study H3K36me2 function. The team found that the overexpression of NSD2 in KMS11 and high levels of H3K36me2 resulted in a significant increase of 5mC genome-wide, especially in the intergenic regions. Consistent with the hypothesis, such intergenic hyper DNA methylation could be specifically reversed by the ablation of T(4;14) translocation. Perhaps more importantly, the team further discovered that the abnormal hyper DNA methylation in KMS11 cells are tumorigenic and that the application of DNA methylation inhibitor, 5-Aza, could effectively suppress the proliferation of KMS11 cells. These findings are exciting, as they raise possibility of applying DNA methylation inhibitor (a FDA proved drug) in treating multiple myeloma bearing T(4;14)/NSD2 translocation.
It is also worth mentioning that during the preparation and submission of this paper, Chao Lu’s team at Columbia University published an article in Nature, entitled “The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape” in September 2019. This other article also revealed a similar finding that H3K36me2 plays a recruitment function for DNMT3A and maintains DNA methylation in intergenic regions. However, the Nature article did not report the stimulation of DNMT3A enzymatic activity by H3K36me2. In additional, they used loss-of-function disease models in the functional studies, therefore did not find potential therapeutic advantage of DNA methylation drug in treating T(4;14) multiple myeloma.
The crosstalk between DNA methylation and histone modification has always been an active area of epigenetics researches. Dr. Lan’s team is one of the first to discover the mechanism of H3K36me2 in regulating intergenic DNA methylation, and is the first to discover the stimulatory effect of H3K36me2 on DNMT3A activity. However, the genome is so complicated that it still remains unclear how different mechanisms work in different regions in regulating DNA methylation and how DNA methylation in different environments affects genome function. These questions call for future studies.
Full text link:https://doi.org/10.1007/s13238-019-00672-y